
1.0 PURPOSE:
SOP for handling of deviation is to establish a system for identification, recording, evaluation and approval of any Deviations from the standard procedure.
2.0 SCOPE:
This SOP for handling of deviation is applicable at (Company name).
3.0 DEFINITIONS:
3.1 Deviation: A Deviation is an activity performed differently and/ or modified than that specified in an approved document that has the potential to affect the safety, identity, strength, quality or purity of a raw material, in-process material or drug product, covering a specified period of time or a specified number of batches.
3.2 Planned Deviation: Any Deviation in the procedure, process, equipment, standard or batch size which is planned, documented, assessed for its impact on product quality and authorized in advance is termed as planned deviation. It is considered as one time activity and is not permanent or long term.
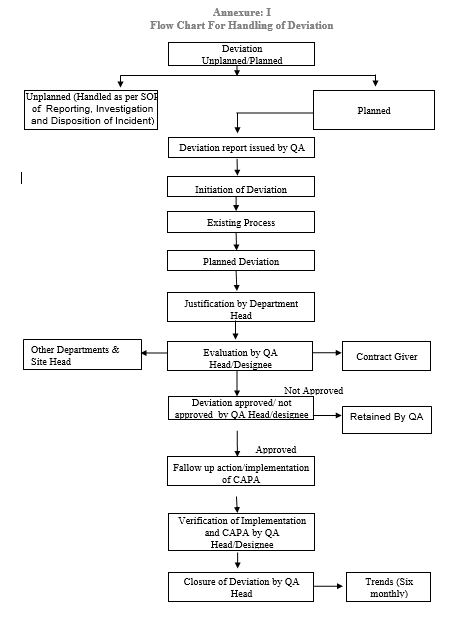
3.3 Unplanned Deviation: While carrying out day to day activities, there are chances of unplanned deviations and events to occur. These deviations are unforeseen, accidental and shall be due to several reasons and shall be handled as per the SOP Reporting, Investigation and Disposition of Incidents.
4.0 RESPONSIBILITY:
4.1 The initiator shall identify the deviation if any, and inform to the department head. To fill the report and follow up. To implement the deviation after approval.
4.2 The Department Head / Designee shall be responsible for justification / evaluation of deviation. To assist QA in investigation.
4.3 Head QA/Designee shall be responsible for evaluation, approval and closure of Deviation. To evaluate the reason and extent of deviation, decision for impact assessment from other departments and review after implementation. To initiate the investigation of the deviation (if required) with the head. Decisive role for approval, rejection of deviation.
5.0 PROCEDURE:
5.1 Unplanned deviation:
5.1.1 The unplanned deviation shall be considered as an incident and handled as per SOP of Reporting, Investigation and Disposition of Incident.
5.2 Planned deviation:
5.2.1 In case of planned deviation the originating department person shall identify the deviation & fill the respective details in deviation Report as per the Annexure-II.
5.2.2 Deviation Report shall be issued by QA to the concerned department on request (Refer Annexure-II).
5.2.3 The QA shall assign a number to the Deviation and enter that number and details of deviation on the “Deviation logbook” as per Annexure-III.
5.2.4 The Deviation number shall be given as
DXX/YYY/ZZZ/AA
Where,
D – Represent the deviation
XX – represents the first two alphabets of company name.
YYY- is department code. For example: Production (PD), Quality assurance (QA), Quality control (QC) etc.
ZZZ- is serial number commencing at 001 for each department in the calendar year.
AA- is last two digits of calendar year.
5.2.5 After receiving the deviation report, originator shall write the general information e.g. Product / Instrument / material name, respective Batch No. / Equipment No., Date of Mfg., Quantity involved & stage.
5.2.6 The originator shall also write the existing system / procedure/ specification/ parameters and proposed deviation for the same. Filled deviation form shall be forwarded to the department head for justification.
5.2.7 Department head shall provide the justification for the proposed deviation and same shall be forward to QA Head / Designee.
5.2.8 QA Head / designee shall review the reason for deviation & its potential effect on the quality of the product / validations/ regulatory status. QA Head / Designee shall comment about requirement of additional studies, validations, stability study and Training.
5.2.9 QA Head/Designee shall give the comments on Deviation and suggest whether separate CAPA required or not.
5.2.10 QA Head/Designee shall decide whether communication to contract giver is required and comment accordingly.
5.2.11 QA Head/Designee shall forward the Deviation to contract giver if applicable.
5.2.12 QA Head/Designee shall identify the concern departments and forward the deviation report to concern departments and Site Head for the comments. Site Head shall evaluate on the financial Implications, if any.
5.2.13 Head QA / Designee shall approve / Not approved the deviation based on evaluation by QA, established procedures, Comments by concerned departments, comments by contract giver, comments by site Head, trend and cGMP requirements.
5.2.14 After approval of deviation concerned department head shall carry out implementation of Corrective action and preventive actions as per the SOP for CAPA and follow up. Department head shall record detail about follow up action.
5.2.15 In event if the deviation is not closed within 30 working days from date of initiation, the concerned department head shall provide justification for the same along with the time line to close the deviation in Annexure-IV.
5.2.16 If preventive action is planned in phased manner, the deviation report shall remain open till the preventive action(s) is completed.
5.2.17 QA Head / Designee shall verify the details of follow up action / corrective and preventive action / documents closed. The QA Head shall verify the details and close the deviation.
5.3 It shall be ensured that if any deviation is associated with a batch then the corrective action and impact assessment of Deviation must be completed before release of the batch.
5.4 If planned deviation is required to be extended for more than once or for longer time or if any change proposed as a result of corrective and preventive action after planned deviation, the same shall be done through the SOP on Change Control. The same shall be mentioned in the deviation report.
5.5 Any conditional approvals involved in planned deviation, evaluation of conformance to be done at the time of batch release.
5.6 If the deviation is planned for a batch it shall be ensured that the action committed is not extended beyond that batch.
5.7 Completed Deviation report shall be filed by QA Department.
5.8 A trend of deviation should be made every six month by Quality Assurance department.
6.0 ABBREVIATIONS:
6.1 QA – Quality Assurance.
6.2 e.g. – Example.
6.3 CAPA – Corrective and Preventive Action
6.4 SOP – Standard Operating Procedure
6.5 cGMP – Current Good Manufacturing Practices
7.0 REFERENCES:
7.1 SOP of “Change Control Programme”
7.2 ICH Q7A: Good Manufacturing Practice for Active Pharmaceutical Ingredients.
7.3 21 code of Federal Regulation, Part 210 and Part 211.
7.4 Rules and Guidance for Pharmaceutical Manufacturers and Distributers, MHRA.
8.0 ANNEXURES:
8.1 Annexure-I: Flow Chart for Deviation Report
8.2 Annexure-II: Deviation Report
8.3 Annexure-III: Deviation Log book
8.4 Annexure-IV: Interim Report for Deviation








9 thoughts on “SOP for Handling of deviation”
Pingback: SOP for management review meeting -
Pingback: SOP for Change control programme -
Pingback: SOP for Responsibilities of the Quality units -
Pingback: 6 Steps of Failure Mode Effects Analysis (FMEA) -
Pingback: SOP for testing and release of in-process, semi-finished and finished product samples -
Pingback: Sop for handling of temporary change control -
Pingback: SOP for Change control programme in pharma -
Pingback: SOP for acceptable quality level (AQL) -
Pingback: SOP for stability studies of finished products -