
1.0 PURPOSE:
To define comprehensive approach to the validation of cleaning procedure for equipment area used in manufacturing process.
2.0 SCOPE:
This SOP for cleaning validation in pharma is applicable to cleaning validation at (Company name).
3.0 DEFINITION:
Cleaning validation: The process of providing documented evidence that the cleaning method employed within a facility consistently control potential carryover of products and extraneous material to the predetermined levels.
Marker Product: Product (worst case) which is identified for cleaning validation in the matrix and as per scoring.
4.0 RESPONSIBILITY:
Engineering: To provide surface areas of the equipment (or) surface areas of equipment’s directly received from manufacturer.
Production: To perform the cleaning of equipment by operators.
Quality Control: To analyze the samples.
Quality Assurance: To check the equipment for cleanliness, collection of samples in coordination with QC and documentation.
5.0 PROCEDURE:
5.1 Cleaning validation approach:
5.1.1 Cleaning validation shall be done in such a way that if the number of product is less, then the approach for individual product cleaning validation is followed. When product get increased and become difficult to validate individually then worst case marker product selection as per product cleaning validation matrix and scoring from the whole product line shall be applicable.
5.1.2 Cleaning validation matrix shall be prepared for manufacturing and packaging lines separately and accordingly protocols and reports shall be prepared separately.
5.1.3 For any new unit/line of products cleaning validation has to be done for first product irrespective of scoring. Upon addition of next products should follow the worst case approach.
5.1.4 For any new unit/line of products, after completion of cleaning validation cleaning verification shall be performed for whichever is applicable.
5.2 Cleaning procedures:
5.2.1 Equipment’s, which are to be considered for cleaning validation, shall be identified.
5.2.2 The following equipment parameters shall be identified before proceeding for cleaning
Validation.
7.2.2.1 Equipment surface area
7.2.2.2 Difficult to clean areas
7.2.2.3 Ease of cleaning and disassembly
7.2.2.4 Physical checking of the areas difficult to clean.
7.2.2.5 Tools used for cleaning.
7.2.3 The equipments shall be cleaned as per the respective SOP for cleaning.
7.2.4 The cleaning details shall be recorded in the respective equipment logbooks.
5.3 Sampling procedure:
5.3.1 Sampling plan for cleaning validation study shall be defined in the cleaning validation protocol.
5.3.2 Sampling shall be done for all the equipment present in the processing line as per cleaning validation protocol irrespective of usage if they are under same AHU (used and unused). Before sampling ensure that the AHU of the area is under operation.
5.3.3 After cleaning QA person along with production personal shall visually inspect the area and equipment before sampling. Swab sampling shall be carried out only if the area and equipment are thoroughly cleaned upon visual checking.
5.3.4 Sampling of swabs for chemical and microbial analysis shall be carried out as per the Respective Protocol/SOPs.
5.3.5 The following parameters shall be considered during sampling:
7.3.5.1 Selection of sampling points in each equipment (Priority to be given for difficult to clean areas).
7.3.5.2 Use of 10 ml medium for swab sampling.
7.3.5.3 Surface area for swab sampling shall be considered as 100 cm2 for chemical analysis and 25 cm2 for microbial analysis.
5.3.6 Wherever rinse sampling (where unable to collect the swab samples) is required for specific equipment, the procedure for the same shall be elaborated in the cleaning validation protocol.
5.3.7 For the sampling points which have perforation (for example sieve of sifter, Coating pan etc.), sample shall be collected 10 x 10 cm2 area in duplicate. Duplicate sampling shall be done from adjacent area of original sampling by using another swab which to be dipped together in diluent to complete 200 cm2 area. This practices shall be followed considering the mesh contains 50 percent of the area of total surface.
5.3.8 The swabs for chemical analysis shall be collected by QA personnel and microbial analysis shall be collected by personnel from micro division under supervision of QA. The sampling details shall be recorded in the sampling plan as per respective cleaning validation protocol.
5.3.9 The collected samples shall be sent to QC along with the sampling plan and sampling advice sheet.
5.4 Analytical method:
5.4.1 Analytical method shall be developed in such a way that LOQ of the method developed shall be less than the limit of the product decided as per the cleaning validation protocol. If the product is site transfer product, then the method validation data originated in the transferring site can be considered for recovery, LOD and LOQ values. If the product is new product then method validation to be carried out.
5.4.2 Analytical method validation shall be carried out for the product identified in the matrix, which includes
5.4.2.1 % Recovery
5.4.2.2 Limit of detection
5.4.2.3 Limit of quantification
5.4.2.4 Accuracy of method
5.4.2.5 Reproducibility
5.4.2.6 Stability of solution.
7.4.3 Analysis of samples shall be carried out as per the defined procedure given in individual
General Testing Procedure referred by individual validation protocol and details shall be recorded.
5.5 Acceptance Criteria:
5.5.1 The following approaches shall be considered for setting acceptance criteria and analytical method limitation.
5.5.1.1 Visual criteria:
Equipments shall be visually cleaned and no traces of previous product residue should appear on equipment surface.
5.5.1.2 Dose criteria:
Not more than 1/1000th dose of the previous product in Maximum Daily Dose (MDD) of next product.
5.5.1.3 10 ppm criteria:
Not more than 10 ppm of any product will appear in another product.
5.5.2 When existing commercial products in the processing line is less and individual product cleaning validation is possible then cleaning validation is carried out individually and limit so obtained for individual product shall be considered for the individual cleaning validation.
5.5.3 In case of worst case approach, a marker product is selected based on worst case senerio form the existing commercial products in the processing line and the limit so obtained shall be considered for the cleaning validation.
5.5.4 The limit is obtained based on the dose criteria/ l0 ppm criteria from the matrix and the product is identified based on the solubility, Therapeutic dose and Toxicity. More than one product may be selected as worst case.
5.5.5 The following points shall be considered for cleaning validation matrix and also in case of product containing more than one API (active Pharmaceuticals ingredient)
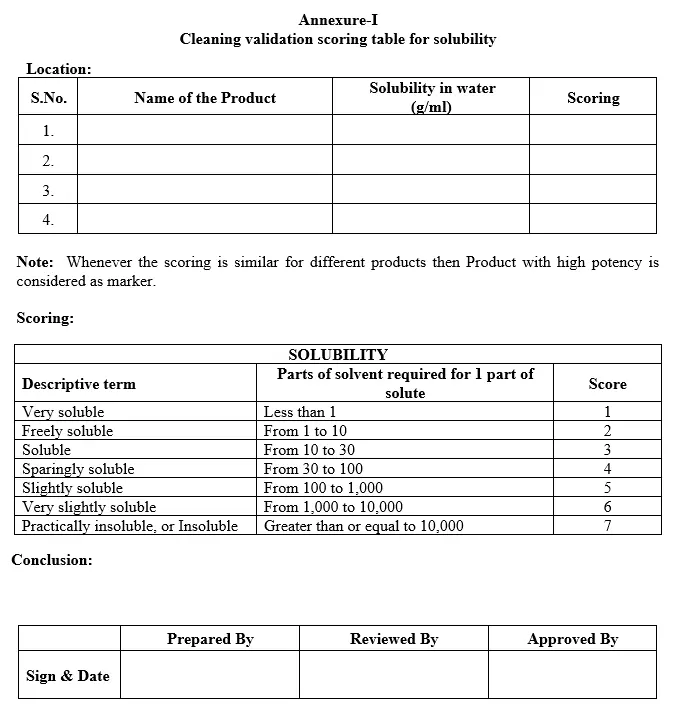
5.5.5.1 Solubility
5.5.5.2 Strength
5.5.5.3 Toxicity
5.5.5.4 Maximum Daily Dose (MDD)
5.5.5.5 Batch size
5.5.5.6 Total surface area of equipments
5.5.5.7 Product list
5.5.5.8 Lower composition among the ingredients (Active and Color).
5.5.6 Matrix shall be prepared for the existing commercial products in the module/line and the limit so obtained shall be considered for the cleaning validation.
5.5.7 When a new product (Exhibit and Commercial) is included, check for the change in marker product status and the limit. If any change is observed in the acceptable limit and marker product, then the below cases apply.
Case-I:
If there is no change in acceptable limit and the marker product, then existing cleaning methods are valid. Hence there is no need of cleaning validation.
Case-II:
If there is a change in acceptable limit and no change in the marker product, then check for the LOQ details and the previous cleaning validation results. If the previous marker product results are failing with new limit, then the existing cleaning methods shall be reestablished and once again cleaning validation shall be repeated.
Case-III:
If there is no change in acceptable limit and there is a change in marker product, then cleaning validation shall be performed for the new marker product.
Case-IV:
If there is change in acceptable limit and change in marker product, then cleaning validation shall be performed for the new marker product.
Case-V:
When new marker Product is identified from the matrix then validation to be performed for new marker product and existing marker product.
5.5.8 When a new equipment (Exhibit and Commercial) is included, check for the change in marker product status and the limit due to change of surface area. If any change is observed in the acceptable limit and marker product, then the below cases apply.
Case-I:
If the new equipment installed is a replacement of existing equipment with same operating principle but small equipment capacity then the total surface area is considered same resulting no change in acceptable limit, then existing cleaning methods are valid. Hence there is no need of cleaning validation.
Case-II:
If the new equipment installed is a replacement of existing equipment with same operating principle but larger equipment capacity but if the increase of surface area is within the range of 3% additional surface area then the total surface area is considered same resulting no change in acceptable limit, then existing cleaning methods are valid. Hence there is no need of cleaning validation
Case-III:
If the equipment installed is a new equipment with same or different operating principle but larger equipment capacity and the increase of surface area is not within the range of 3% additional surface area then the total surface area will changed, resulting change in acceptable limit, In such case check for the LOQ details and the previous cleaning validation results. If the previous marker product results are failing with new limit, then the existing cleaning methods shall be reestablished and once again cleaning validation shall be repeated.
5.5.9 When a cleaning validation is performed for a single module is needs separation of modules due to use of different technique in new formulation. Eg if cleaning validation is performed for dry granulation and compression & coating together and when a new formulation is introduced with wet granulation, compression and coating. Then due to introduction of new formulation the cleaning validation module needs separation. Then matrix shall be prepared for dry granulation, wet granulation and compression differently. In such case, the below cases apply.
Case-I:
If the new product used wet granulation technique then the existing product validation for dry granulation compression and coating shall be considered for dry granulation and no separate cleaning validation is required for dry granulation. The same shall be concluded in cleaning validation matrix.
5.6 For Visual criteria:
5.6.1 Equipment shall be checked for the cleanliness visually.
5.6.2 Minimum of two persons shall check for the equipment cleanliness.
5.6.3 Visual checking shall be checked in angled position and in flat position wherever applicable.
5.7 For Rinse water sample:
5.7.1 Rinse sample shall be collected for the equipments, which are difficult to clean and difficult to collect the sample for analysis as per respective protocol.
5.7.2 After cleaning of those equipment’s, rinse the equipment with one liter of purified water or as mentioned in the respective process area cleaning validation protocol and collect the rinse water into a cleaned SS vessel.
5.7.3 Collect 10 ml of sample from the rinsed water and send for chemical testing. Analyze the samples as per the respective cleaning test procedure mention in the respective product cleaning validation protocol.
5.7.4 Quality Control person shall carryout analysis as per the test procedure or reference specification number given in concerned cleaning validation protocol and write the details in Sampling plan.
5.7.5 Head – QC or designee shall check the details and approve the samples upon confirmation the results are within specified acceptance criteria as mentioned in the cleaning validation protocol.
5.7.6 Acceptance criteria for rinse water for chemical analysis shall be considered as per the respective process area cleaning validation protocol. Maximum allowable carryover for rinse water per equipment in mcg is calculated as below.

5.8 For Microbial swab sample:
5.8.1 Microbial swabs shall be collected after cleaning the area and equipment’s.
5.8.2 Collected Microbial swab samples shall be sent for Microbial testing and analyze the samples as per the respective microbial swab testing procedure/SOP mention in the respective product cleaning validation protocol.
5.9 For Chemical swab sample:
5.9.1 Dose criteria:
5.9.1.1 For dose criteria, product matrix shall be prepared and to be reviewed whenever there is inclusion of new molecule in the product line.
5.9.1.2 Acceptance criteria shall be calculated as per Dose criteria, l0 ppm criteria. Minimum value obtained between these three criterions shall be selected as limit.
5.9.1.3 Cleaning validation matrix shall be prepared and to be revised whenever there is inclusion of new drug in the product line.
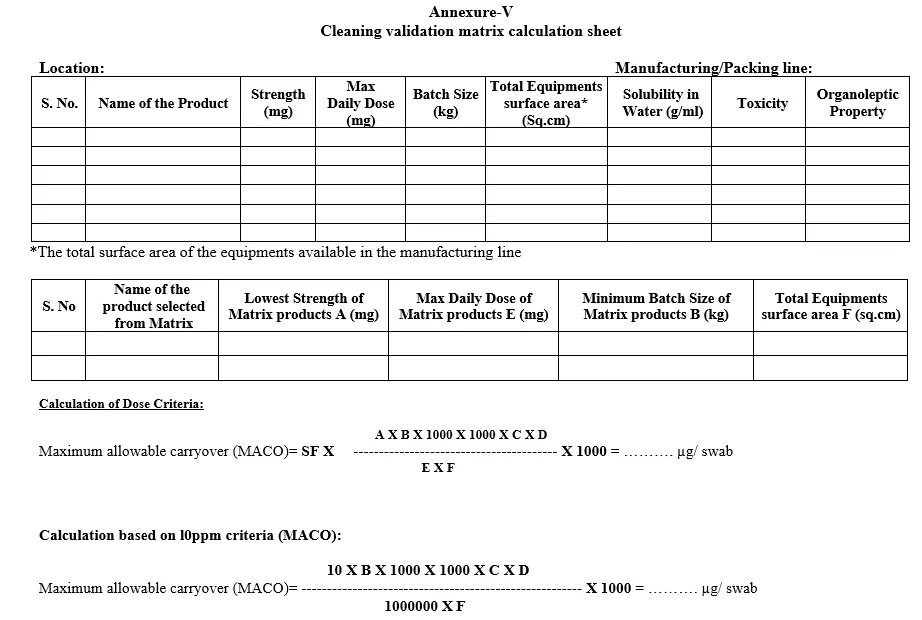
5.9.1.4 The acceptance limit for maximum allowable residue is 1/1000th of the lowest strength of the product and maximum daily dose of the product (active drug) in the matrix. Considering the smallest batch size, maximum daily dose and equipment surface area of products in matrix, the limit of residue is calculated as below:
5.9.1.5 Calculation of Dose Criteria:

5.9.2 10 ppm CRITERIA:
Calculation of maximum allowable residue cannot be done based on dose criteria when subsequent product to manufacture not known, Hence calculation of maximum allowable residue shall be done based on 10 ppm criteria. In case dose criteria calculation of maximum allowable residue found more than 10 ppm, calculation of maximum allowable residue base on 10 ppm criteria.

5.10 Cleaning validation protocol and report:
5.10.1 Common cleaning validation protocol shall be prepared for each process area based on worst case product. Each process line shall be separated based on manufacturing process (e.g. Wet granulation and dry granulation both are separate process hence separate protocol is required with separate selection of worst case). The protocol heading should indicate the process area. The limit obtained through SOP shall be used in the validation protocol.
6.0 ABBREVIATIONS:
SOP : Standard operating procedure
QA : Quality Assurance
GMP : Good Manufacturing Practice
LOQ : Limit of quantification
SS : Stainless steel
MDD : Maximum daily dose
LOD : Limit of detection
Ppm : Parts per million
AHU : Air handling Unit
mcg : microgram
MACO : Maximum allowable carryover
7.0 ENCLOSURES:
Annexure-I: Cleaning Validation Scoring table for solubility
Annexure-II: Cleaning Validation Scoring table for Therapeutic dose
Annexure-III: Cleaning Validation Scoring table for Toxicity
Annexure-IV: Cleaning Validation total Scoring table
Annexure-V: Cleaning Validation Matrix Calculation Sheet




Also read: Cleaning Validation protocol
Also read: SOP for process validation of pharmaceutical products