
1.0 PURPOSE:
2.0 SCOPE:
2.1 This procedure for change control programme in pharma is applicable to (Company name).
2.2 This procedure is applicable to all changes but not limited to process steps, procedures, documents, specifications, raw materials, facilities, utility, Support systems, equipment’s, instruments, packaging materials, product labels, computer software and Vendor/Supplier (if any) which has an impact on Quality systems and Product Quality.
3.0 DEFINITIONS:
3.1 Change control:
A formal system by which qualified representatives of appropriate discipline review proposed or actual changes that might affect a validated status. The intent is to determine the need for action that would ensure and documents that the system is maintained in a validated state.
3.2 Initiator:
Any member of staff who identifies a need for change. Any employee of organization can complete the change request form for processing as per SOP.
3.3 Major Change:
A change that has a substantial potential to have an adverse effect on the identity, strength, quality, purity or potency of a drug product as these factors may relate to the safety or effectiveness of the drug product.
A change resulting Qualification /Re qualification of area /Equipment
A change affecting multiple departments/facilities/equipment train.
Following may be classified into Major, but not limited to:
3.3.1 Facility related changes (Expansion and modification of facility)
3.3.2 New product introduction in facility.
3.3.3 Change in Formula/batch size/manufacturing process.
3.3.4 Change of Source of raw materials and primary packaging material.
3.3.5 Changes in Computer Software/Hardware –Major parts.
3.3.6 Change like marketing authorization variation
3.3.7 Installation of new equipment (Compression machine, Coating machine, Fluidized bed dryer (FBD) Blister packing machine, Filling machine ,Lyophilser ,Steam sterilizer etc..)
3.3.8 Utility related changes (Water system, Compressed air etc.)
3.3.9 Change in sterilization parameters /Change in sterilization filter size (i.e. 6” to 4” etc.)
3.3.10 Change in labeled storage condition.
3.3.11 Extension of expiry date of finished product based on new / revised stability or on full time self-life data.
3.4 Minor Change:
A change, which does not have any impact on the Quality, purity, strength and integrity of a drug product.
A change, which does not result in Qualification /Re qualification of area /Equipment.
A change does not affect multiple departments/facilities/equipment train.
Following may be classified into Minor, but not limited to:
3.4.1 Change in document number.
3.4.2 Revision of SOP or documents based on validation /qualification recommendation.
3.4.3 Equipment (Same make, model, and working principle) addition in BMR & BPR.
3.4.4 BMR revision after process validation recommendation.
3.4.5 Shifting of Non grounded equipment (like Sifter, Roll compactor & Multimill etc.)
3.4.6 Installation of New shipper weighing balance, Multimill, Solution preparation vessel & Peristaltic pump, Hardness Tester & Shrink wrapping machine.
3.4.7 Change in color Gowning etc.
3.4.8 Note: Examples are provided for understanding only. Actual classification shall be done with appropriate impact assessment.
4.0 RESPONSIBILITY:
4.1 Initiator:
Shall be responsible for
4.1.1 Initiation of Change request form.
4.1.2 Filling up of Change request form (PART A).
4.1.3 Proposing justification for the change.
4.1.4 Obtaining comments/concurrence from the concerned based on QA evaluation.
4.1.5 Returning the complete form to QA.
4.2 Department Head / Designee:
Shall be responsible for :
4.2.1 Implementation of the change.
4.2.2 Review of implemented changes.
4.2.3 Ensuring the change control is closed.
4.3 Manager QA/Designee:
Shall be responsible for:
4.3.1 Categorization of change.
4.3.2 Check the adequacy of the justification provided.
4.3.3 Review the impact of the changes.
4.3.4 Review the impact of documents affected due to the changes.
4.4 Head Regulatory / Designee:
Shall be responsible for:
4.4.1 Any impact on the regulatory dossiers.
4.4.2 Evaluate the change control with regulatory perspective.
4.5 Head QA / Designee:
Shall be responsible for:
4.5.1 Review the change proposal and evaluation.
4.5.2 Communication with the contract giver/Regulatory department.
4.5.3 Evaluation of impact on quality.
4.5.4 Verification of change implementation.
4.5.5 Approval / Rejection of change proposed.
4.5.6 Closure of Change Control.
5.0 PROCEDURE:
5.1 A change to any of the following shall be subjected to Change control but not limited to,
5.1.1 Changes in Facility:
5.1.1.1 Facility Design/Facility Layout/Engineering Drawings.
5.1.1.2 Change in Location or Addition/Alteration of manufacturing site.
5.1.1.3 Change in HVAC Parameter.
5.1.2 Changes in Equipment:
5.1.2.1 Major changes in the Existing Equipments.
5.1.2.2 Major changes in the Existing Instruments.
5.1.2.3 Calibration tolerances (or) Frequency of calibration.
5.1.2.4 Critical utility such as water, steam, filtered air supplies, power, vacuum systems.
5.1.2.5 Any change in the frequency of preventive maintenance or calibration.
5.1.2.6 Transfer of equipments from one location to another location.
5.1.2.7 Change control is not required in case of portable equipments & Balances transfer from one location to another location within the department.
5.1.3 Changes in software:
5.1.3.1 Change control is not required for the up gradation of Patches and service pack however change control shall be raised in case of version change.
5.1.4 Changes in Process:
5.1.4.1 Addition/Deletion/Modification of RM/PM (or) quantity in manufacturing/Packing Process.
5.1.4.2 Any changes in Manufacturing process (Method of manufacturing).
5.1.4.3 Any change in Batch size and storage conditions of a product.
5.1.4.4 Any change in the cleaning method and cleaning procedures.
5.1.4.5 Any changes made after process validation.
5.1.4.6 Any changes in materials, vendor and specifications as per regulatory requirements.
5.2 Before initiating a process change Deviation shall be raised as per the current version of the procedure for “Handling of Deviation” for three consecutive batches.
5.2.1 Before initiating the change control for the process the following shall be monitored for three consecutive batches.
5.2.1.1 All in process parameters trend.
5.2.1.2 All Process Quality parameters trend.
5.2.1.3 All Testing Parameters Trend.
5.2.1.4 Trend for Consistent Yield.
5.2.1.5 Long term/Accelerated Stability studies shall be carried out for at-least one of the batches to confirm that it remains within the specified limits.
5.2.1.6 If all the above are found within the limits the change control shall be initiated and approved by assuring that the changes are reflecting in all relevant documents/procedures and Batch manufacturing records.
5.2.2 Changes in Materials:
5.2.2.1 Any change in Raw materials (Active/ Excipients)
5.2.2.2 Any change in Packing Materials.
5.2.2.3 Any changes in quantity of materials.
5.2.3 Changes in Testing (Material and Finished Product):
5.2.3.1 Any changes required in specifications due to change in Pharmacopoeia.
5.2.3.2 Product/Material Specifications (Addition/Deletion of any tests).
5.2.3.3 Any changes in Method of Analysis.
5.2.3.4 Any changes in Stability testing and Stability Protocols.
5.2.3.5 Any changes made in the specifications due to trend/Annual product Quality Review.
5.2.3.6 Any changes due to planned deviations as a part of Continuous improvement.
5.2.3.7 Any changes in the Self-life of the product.
5.2.4 Changes in Documents:
5.2.4.1 Any changes as per cGMP regulations.
5.2.4.2 Any changes in MFC or MPC / Batch Manufacturing Documents/Master Documents
5.2.4.3 Any changes made in Quality systems SOPs/STPs/GTPs/ Specifications.
5.2.4.4 Any changes in In- process /Finished product Labels.
5.2.4.5 In case of new document preparation, change control need not be raised.
5.2.4.6 In case of any document to be made obsolete, change control shall be raised along with justification/reason.
5.2.4.7 In case of shifting of any SOP from one department to other.
5.3 This procedure shall provide a formal change control system so as to prevent unauthorized and unreported changes to established facility systems and processes.
5.4 Change control Initiation:
5.4.1 Concerned department shall initiate change request through Annexure-I (Change Request Form).
5.4.2 Change Request Form (CRF) shall be issued by QA to the concerned department on request after assigning a specific change control number.
5.4.3 The Change control number shall be given as
CC/YY/ZZZ/AA
Where,
CC – represents change control.
YY- represents first two alphabets of the Company name.
ZZZ- is serial number commencing at 001 for each department in the calendar year.
AA- is last two digits of calendar year.
5.4.4 The QA shall assign a number to the Change Request Form and enter that number and proposed change on the change control request logbook (Annexure-II).
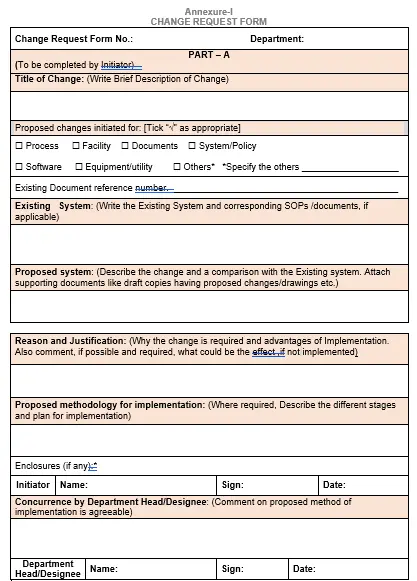
5.5 Part A of the change request Form:
Write the details in the part a as follows:
5.5.1 Department: Write the name of concerned department.
5.5.2 Title of change: Write brief description of change.
5.5.3 Change initiated for tick mark the right choice (s) from process, facility, documents or others depending on the change.
5.5.4 Existing document reference number: Write the number of the documents to be changed.
5.5.5 Changes proposed and justification: Write the existing system, proposed system in the space provided. In addition, Provide the adequate justification and proposed methodology for implementation of the change proposed. Attach supporting data, documents or drawing in support of the change proposed or provide cross reference to documents wherever applicable.
5.5.6 Enclosures: Provide a list of enclosures attached to support the change including approved deviation forms, communications etc. A copy of the proposed revised document can be attached to the proposal, if required.
5.5.7 The Initiator shall take concurrence by department Head/Designee.
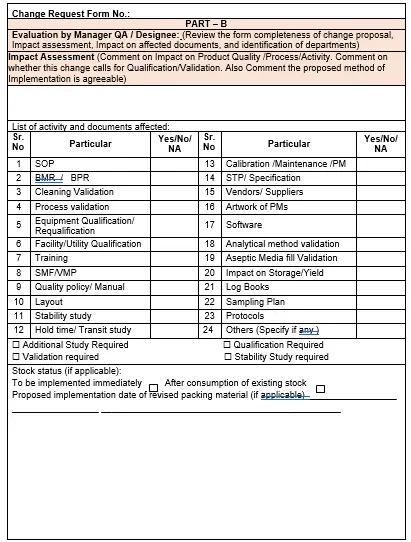
5.6 Part B of Change Request Form:
5.6.1 The initiator shall send the proposal for evaluation by Manager QA/ Designee.
5.6.2 Manager QA/ Designee shall evaluate the Impact assessment of the change with respect to (Not limited to).
5.6.3 Impact on quality of product regarding in-house specifications and specific customer requirements.
5.6.3.1 Impact on stability, impurity profile.
5.6.3.2 Impact on test limits, statistical testing parameters.
5.6.3.3 Documents needed to be amended with respect to change.
5.6.3.4 Requirement of addition studies, validation and / or qualification.
5.6.3.5 Adequacy of justification and supporting documents.
5.6.3.6 Impact on performance of method of analysis.
5.6.3.7 Impact on other products manufacturing in the same facility.
5.6.3.8 Impact on supply chain/Material procurement.
5.6.3.9 Impact on current validation status and validation requirements.
5.6.4 Manager QA/ Designee shall list the activities to be performed and documents affected by the change.
5.6.5 Manager QA/ Designee shall evaluate the list of activities, documents affected and categorize the change proposed as Minor or Major category depending on the nature and degree of changes.
5.6.6 After completion of evaluation by QA as per the above assessment, Manager QA/Designee shall identify the concern departments affected by the proposed change and forward the change control form with his comments to concern departments for their review and comments.
5.7 Part C of Change Request Form:
5.7.1 Each affected department identified shall review and the Comments made on the proposed change.
5.7.2 If there is no consensus in the departments on change proposal, Head QA shall constitute change control committee and discuss on the concerns raised. The committee shall have Head of initiating department, other affected departments and any other departments or SMEs (Subject matter experts) as members. Head QA is authorized to select the committee members.
5.7.3 The initiator shall obtain concurrence of QA Head, in which QA Head also state the requirements of communication to contract giver and site head.
5.7.4 Based on proposed change, If there is any Major Financial implications on change the change control shall be Reviewed and approved by the site Head with respect to financial implications on the change.
5.8 Change request form shall be sent for obtaining comments/concurrence of the contract giver (Wherever applicable). Contract giver shall comment and sign in the space provided.
5.9 The change shall be approved only after obtaining concurrence from contract giver, wherever applicable.
5.10 The initiator shall forward the change request form for the approval of Head QA/designee. Head QA/designee shall review the Change control proposal with respect to (not limited to)
5.10.1 Impact on regulatory commitments.
5.10.2 Impact on stability, impurity profile
5.10.3 Requirement of trials, validation and additional testing.
5.10.4 Adequacy of justification and supporting documents
5.10.5 Impact on performance of method of analysis.
5.11 Head QA/Designee shall approve or not approve a change based on the proposal, adequacy of justification provided.
5.12 Proposed changes shall be carried out by initiator, only after the approval of Change control by Head QA/designee.
5.13 Carry out the proposed changes as specified in the change control request form.
5.14 PART D of change request form:
5.14.1 Quality assurance shall list out the documents/process affected and actions required by considering the comments from all departments and Head QA in Part D of Annexure-I.
5.14.2 QA shall update the status of Approval /Rejection in change control log and communicate the same to change initiator and other affected departments for implemetation of change and also to inform the actions required on different documents/process as identified by QA.
5.14.3 Initiator shall implement the change. The status of each activity shall be updated after its completion in the original change control form.
5.14.4 Status of change, Implemetation and effectiveness shall be updated by Head of department.
5.14.5 If the change not implemented or any discrepancies in implementation shall be mentioned along with justification.
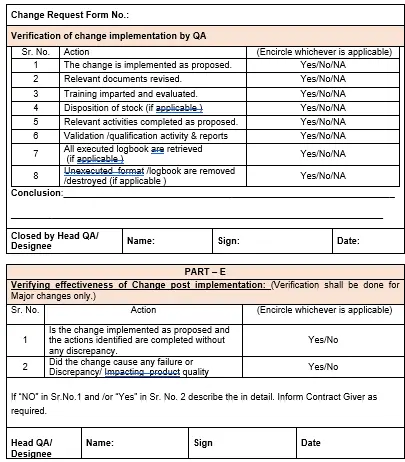
5.15 Verification of change implementation:
5.15.1 After Implementation of change Head QA/ Designee shall check the adequacy of closure on below but not limited to,
5.15.2 Verification of all documents as per the change control request.
5.15.2.1 Verification of training done on changes made as per the change control
5.15.2.2 Validation / Qualification activity and reports.
5.15.2.3 Disposition of Stock (If applicable).
5.15.2.4 Verification of relevant activities completion status.
5.15.2.5 On implementation of the approved change, Head QA/designee shall review the status of implementation of the change against action specified and encircle whichever is applicable along with other comments if any.
5.15.3 On verification of successful implementation of change, Head QA/Designee shall conclude and close the change request form.
Note: Before closing of change control form, all supporting attached documents are stamped as “Reference Doc. No.: ” and write the reference change control No. on each page of attached documents. And after closing of change control put ″CLOSED” stamp on first pages in blue colour.
5.15.4 Verifying effectiveness of change after implementation by QA:
5.15.4.1 QA shall verify the completion of the proposed changes and activities identified. An impact assessment of the implemented change shall be done by QA and documented in Annexure-I.
5.15.4.2 If the approved changes caused any failure or discrepancy, QA shall comment and if required, it shall be notified to contract giver.
5.15.4.3 Verify the effectiveness of change after implementation (i.e. 3 months) shall be carried out only for major changes.
5.16 The original signed and approved / rejected CRF along with supporting documents shall be retained by QA.
5.17 A photocopy of the closed change control shall be given to regulatory affairs for their reference (if required). Regulatory department shall ensure that the regulatory agencies are update of the change by in forming the change details (whenever applicable).
5.18 Time line:
5.18.1 All procedural changes to GMP documents (specification, SOP, BMR and BPR) shall be closed within 30 working days from the date of approval by QA Head.
5.18.2 Equipment and facility related changes should be completed in 6 months. In case of delay, justification shall be provided by initiating department and authorized by Head QA/Designee as per Annexure-III (Interim reports of change control).
NOTE: For all change control form shall be approved within 30 working day from the date of issuance of change control. In case of delay, justification shall be Provided by initiating department and authorized by Head QA/Designee as per Annexure-III (Interim reports of change control).
5.19 Trending of change control:
5.19.1 A summary of the change control shall be prepared half yearly. The summary should include departments, type of change, implementation assessment etc. Summary shall include on effectiveness of implementation of changes also. The summary shall be prepared QA – Executive and shall be reviewed and Signed by Head – QA/Designee.
6.0 ABBREVIATIONS:
6.1 QA :Quality Assurance.
6.2 API :Active Pharmaceutical Ingredient
6.3 CRF :Change Request Form.
6.4 HEPA :High Efficiency Particulate Air
6.5 SOP :Standard Operating Procedure
6.6 STP :Standard test procedure
6.7 GTP :General test procedure
6.8 HVAC :Heating Ventilation and air conditioning
6.9 RM :Raw material
6.10 PM :Packing material
6.11 MFC :Master formulation card
6.12 MPC :Master packing card
6.13 No. :Number
6.14 SMEs :Subject matter experts
7.0 REFERENCES:
7.1 Guidance for Industry – Immediate release solid oral doses forms- Scale up and post approval changes: Chemistry, Manufacturing and control, in Vitro Dissolution Testing and in Vivo Bioequivalence Documentation.
7.2 Guidance for Industry- Change to an approved NDA or ANDA.
7.3 ICH Q7A: Good Manufacturing Practice for Active Pharmaceutical Ingredients.
7.4 21 code of Federal Regulation, Part 210 and Part 211.
7.5 Guidance on variations to a prequalified Dossier (WHO)
7.6 Rules and Guidance for Pharmaceuticals Manufactures and distribution, MHRA.
8.0 LIST OF ANNEXURES:
8.1 Annexure-I: Change Request Form
8.2 Annexure-II: Change Control Request Logbook
8.3 Annexure-III: Interim Report for Change Control.





Also read: Sop for handling of temporary change control
Also read: SOP for Handling of deviation