
1.0 PURPOSE:
To describe the procedure for administration of Corrective and Preventive action (CAPA) , including tracking and reporting of the status of CAPA.
2.0 SCOPE:
The SOP for corrective and preventive action (CAPA ) is applicable to Pharmaceutical Manufacturing Facilities of (Company name). This SOP is also applicable for stability studies, validation studies and analytical studies at Research and development department, being conducted for products intended for regulatory filing.
3.0 DEFINITIONS:
3.1 Corrective action: Action taken to rectify, fix or correct a specific deviation, defects or undesirable situation in order to prevent recurrence.
3.2 Preventive action: Action taken to eliminate the cause of potential nonconformity, defect, or other undesirable situation in order to prevent occurrence.
4.0 RESPONSIBILITY:
4.1 The Head of the department or the designee is responsible to take CAPA where the issue is identified.
4.2 QA personnel shall be responsible for issuance of CAPA Form to concerned department.
4.3 Head – Quality Assurance or the designee to ensure implementation and compliance of this procedure.
5.0 PROCEDURE:
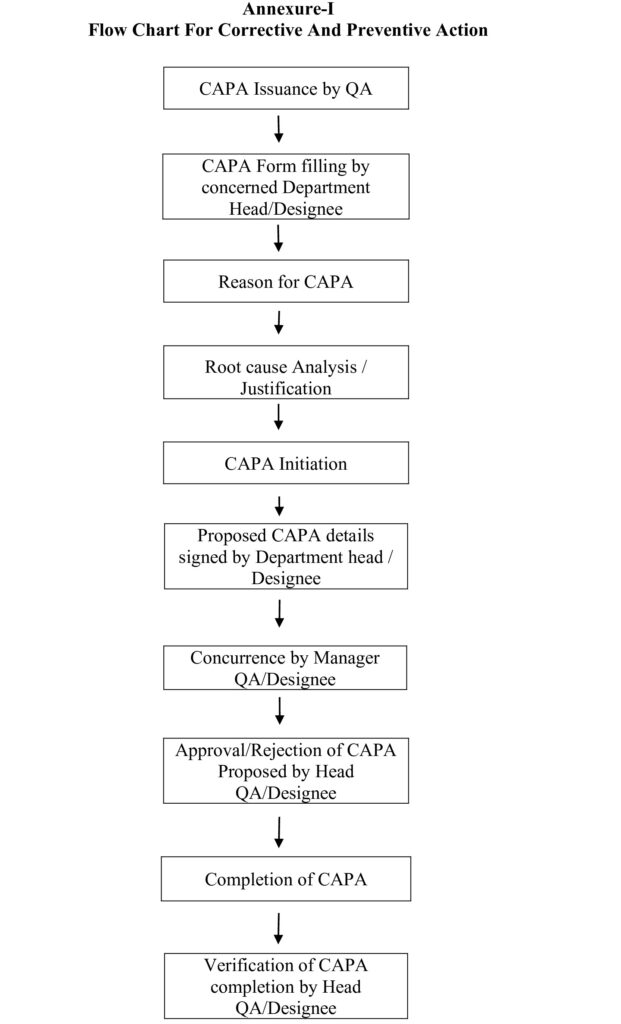
5.1 QA personnel shall issue the format for CAPA as per Annexure-II to the concerned department on request.
5.2 Source documents for (Corrective and preventive action) CAPA includes but not limited to:
5.2.1 Review of BMR, BPR and QC records
5.2.2 Internal audit reports
5.2.3 Deviation
5.2.4 Product failure
5.2.5 Incident Reports
5.2.6 Laboratory investigation
5.2.7 Market complaint
5.2.8 External/Customer audits
5.2.9 Returns goods
5.2.10 Data and Risk analysis related to operations and quality systems
5.2.11 Annual Product Reviews
5.2.12 Regulatory inspection reports
5.2.13 Management action plan
5.2.14 Changes in regulatory / Pharmacopoeia requirements
5.3 Department Head / Designee shall decide the need for CAPA during assessment of any source documents mentioned above.
5.4 CAPA involves the Following but not limited to:
5.4.1 Identification of actual or potential quality or compliance non-conformance.
5.4.2 Evaluation of significance of non-conformance and urgency of any remedial action.
5.4.3 Control of any affected product or material and assessment of the impact on any batches or part of batch of material or product not directly involved.
5.4.4 Implementation of any remedial action.
5.4.5 Investigation into the root cause of actual or potential non-conformances including documenting of results.
5.4.6 Evaluation of the need for action to ensure actual non-conformance do not recur or potential non-conformance do not occur.
5.4.7 Determination of the required corrective and/or preventive action.
5.4.8 The action shall address both the non-conformance and the root cause.
5.4.9 Recording the results of action taken.
5.4.10 Review the corrective or preventive action taken.
5.5 Initiating Department Head / Designee shall write the following details on the format for CAPA:
5.6 Department
5.7 Source document
5.8 Source document number (if applicable)
5.9 Proposed completion date
5.10 Initiating Department Head / Designee shall fill the details like ‘Reason for CAPA’, ‘Root cause analysis / Justification’ with proposed CAPA.
5.11 Initiator shall obtain the concurrence from Manager QA / Designee for the proposed CAPA.
5.12 QA Head / Designee shall Approved/Not Approved the CAPA.
5.13 The CAPA form shall be treated as a tracking form of corrective and preventive action from source document.
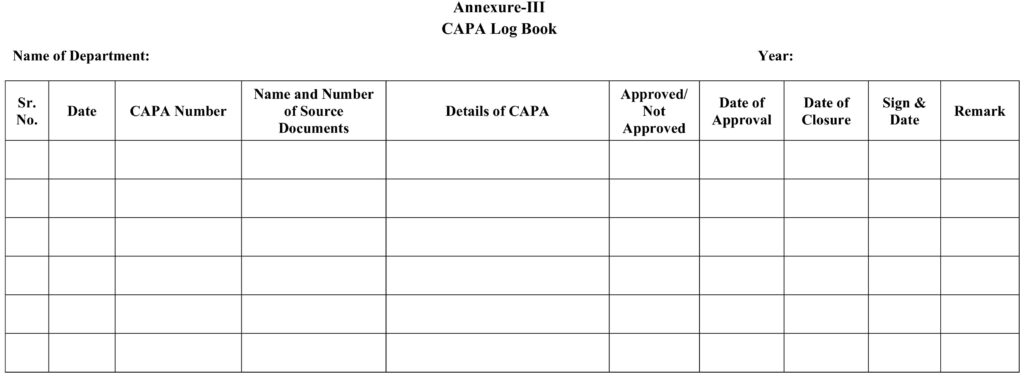
5.14 The record of CAPA issued to initiating department shall be maintained in log of corrective and preventive action as per Annexure-III.
5.15 The chosen corrective and/or preventive action must be progressed according to the target completion date agreed by the respective department head.
5.16 Concerned department head / designee shall carryout a review of CAPA implementation and send the CAPA form to QA Head or Designee for verification.
5.17 QA Head / Designee shall verify the implementation of CAPA with review of supporting documents, as appropriate.
5.18 Any change proposed as a result of CAPA shall be initiated through “Change Control Programme”. Reference of the change request form to be mentioned in the CAPA form, where appropriate.
5.19 If CAPA involve changes to process that may impact previous validation or regulatory approvals, changes should be implemented after verification of compliance with filing / regulatory requirements.
5.20 In case, CAPA is required for product manufactured for a contract giver, acceptance shall be obtained from the contract giver through source documents. Relevant communication shall be attached to source document, as appropriate.
5.21 Copy of completed CAPA shall be provided to concerned department head by QA, if required.
5.22 Department shall compile the CAPA information and submit the summary to the management during management review meeting.
5.23 Information and documents related to CAPA drawn from self inspection, External/Customer Audits and regulatory inspections are considered confidential and can only made available to other than regulatory review when approved by Managing Director or Vice President Operation or Site Head.
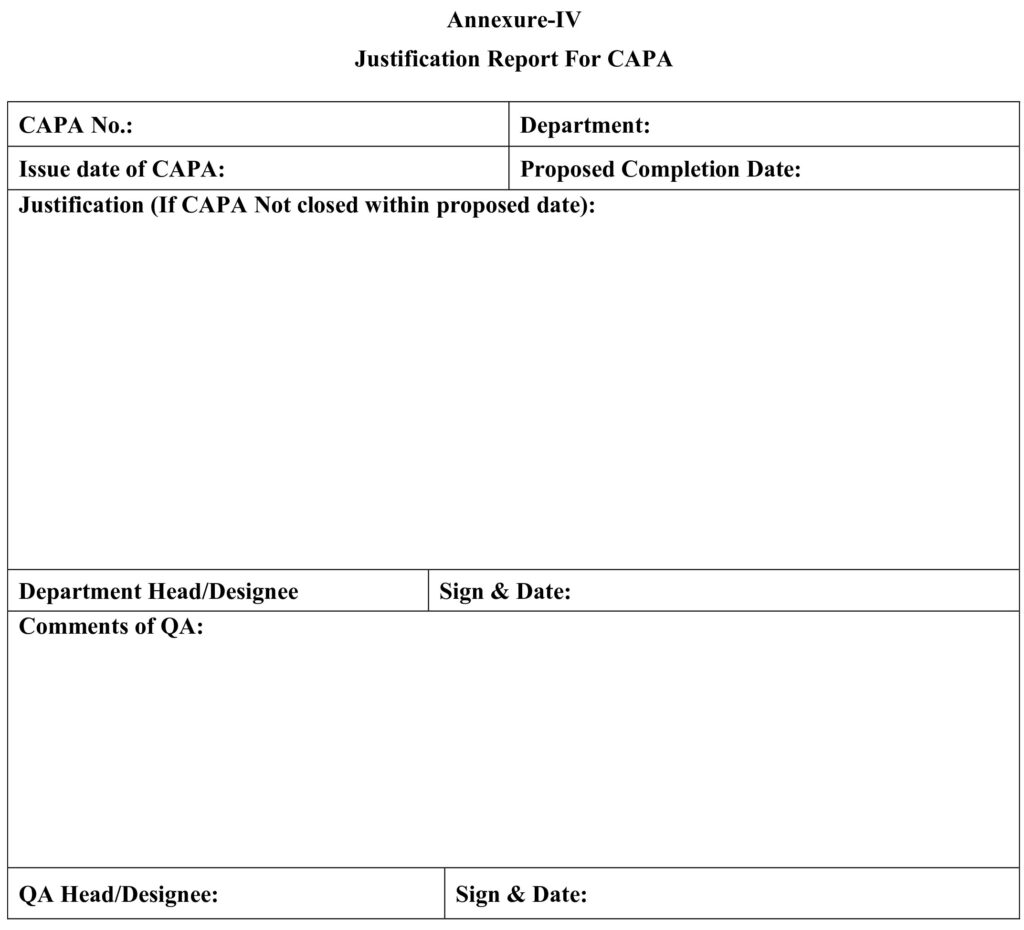
5.24 In event of proposed CAPAs are not completed within proposed date a justification shall be provided by Head of the department and same shall be reviewed and approved by Head QA as per Annexure-IV.
5.25 The CAPA shall be numbered serially in the calendar year as
CXX/YY/ZZZ/AA
Where
C stand for CAPA
XX– are first two alphabets of the Company name or of the name of contract giver.
Incase if the significant two alphabets of the name of two different contract givers are same then first and last alphabets of the name of later shall be considered.
YY- is department code. For example: Quality assurance (QA), Quality control (QC), Production (PD) etc
ZZZ – is serial number commencing at 001 for each department in calendar year.
AA – is last two digit of calendar year.
6.0 ABBREVIATIONS:
6.1 CAPA – Corrective and Preventive Action
6.2 SOP – Standard Operating Procedure
6.3 QA – Quality Assurance
6.4 e.g. – Example
7.0 REFERENCES:
7.1 EU Guidelines for Good Manufacturing Practices for Medicinal Products for Human and Veterinary use, Chapter 1, Pharmaceutical Quality System
7.2 Rules and Guidance for Pharmaceutical Manufacturers and Distributers, MHRA
7.3 ICH Q7A: Good Manufacturing Practice for Active Pharmaceutical Ingredients
7.4 SOP of “Chang Control Programme”
8.0 LIST OF ANNEXURES:
8.1 Annexure –I: Flow Chart for Corrective and Preventive Action.
8.2 Annexure –II: Corrective and Preventive Action Report.
8.3 Annexure –III: CAPA Logbook.
8.4 Annexure –IV: Justification Report for CAPA.


7 thoughts on “SOP for Corrective and Preventive Action (CAPA)”
The article is very informative. Please write an article on handling of deviation.
Pingback: SOP for Handling of deviation
Pingback: SOP for Self-inspection (Internal Audit) -
Pingback: SOP for Handling of Alarms for Critical Equipments -
Pingback: SOP for management review meeting -
Pingback: SOP for Responsibilities of the Quality units -
Pingback: Sop for handling of temporary change control -